MindSponge分子动力学模拟——定义Collective Variables(2024.02)
技术背景
在前面的几篇博客中,我们介绍了
MindSponge分子动力学模拟框架的基本安装和使用
和
MindSponge执行分子动力学模拟任务的方法
。这里我们介绍一个在增强采样领域非常常用的工具:Collective Variable(CV),或者我们也可以直接称呼其为一个物理量。因为像化学反应或者是蛋白质折叠等问题中,经常会存在一个“路径(Path)”,使得反应沿着这个路径来进行。其中最简单的一种形式,就是成键断键。换句话说,我们可以通过调控这根键的键长,进而去调控这其中的化学反应,这也是分子力学层面的增强采样的一个基本思想。而随着增强采样技术的发展,越来越多的形式的CV被应用在不同的领域和问题当中。本文将会介绍,如何在基于深度学习框架MindSpore的分子动力学模拟软件MindSponge中,去定义一个CV。
能量极小化
首先我们使用到前面文章中提到过的MindSponge的能量极小化这个案例作为示例,优化的是这样的一个体系:

对应的pdb文件为:
REMARK Generated By Xponge (Molecule)
ATOM 1 N ALA 1 -0.095 -11.436 -0.780
ATOM 2 CA ALA 1 -0.171 -10.015 -0.507
ATOM 3 CB ALA 1 1.201 -9.359 -0.628
ATOM 4 C ALA 1 -1.107 -9.319 -1.485
ATOM 5 O ALA 1 -1.682 -9.960 -2.362
ATOM 6 N ARG 2 -1.303 -8.037 -1.397
ATOM 7 CA ARG 2 -2.194 -7.375 -2.328
ATOM 8 CB ARG 2 -3.606 -7.943 -2.235
ATOM 9 CG ARG 2 -4.510 -7.221 -3.228
ATOM 10 CD ARG 2 -5.923 -7.789 -3.136
ATOM 11 NE ARG 2 -6.831 -7.666666 -4.087
ATOM 12 CZ ARG 2 -8.119 -7.421 -4.205
ATOM 13 NH1 ARG 2 -8.686 -8.371 -3.468
ATOM 14 NH2 ARG 2 -8.844 -6.747 -5.093
ATOM 15 C ARG 2 -2.273 -5.882 -2.042
ATOM 16 O ARG 2 -1.630 -5.388 -1.119
ATOM 17 N ALA 3 -3.027 -5.119 -2.777
ATOM 18 CA ALA 3 -3.103 -3.697 -2.505
ATOM 19 CB ALA 3 -1.731 -3.041 -2.625
ATOM 20 C ALA 3 -4.039 -3.001 -3.483
ATOM 21 O ALA 3 -4.614 -3.643 -4.359
ATOM 22 N ALA 4 -4.235 -1.719 -3.394
ATOM 23 CA ALA 4 -5.126 -1.057 -4.325
ATOM 24 CB ALA 4 -6.538 -1.625 -4.233
ATOM 25 C ALA 4 -5.205 0.436 -4.039
ATOM 26 O ALA 4 -4.561 0.930 -3.116
ATOM 27 OXT ALA 4 -5.915 1.166 -4.728
TER
那么我们就可以通过调控体系中每个原子的坐标,来使其能量达到一个最低值。相关代码如下所示,这里我们用的是一个MindSpore自带的Adam优化器:
from mindspore import nn, context
from sponge import ForceField, Sponge, set_global_units, Protein
from sponge.callback import RunInfo, WriteH5MD
# 配置MindSpore的执行环境
context.set_context(mode=context.GRAPH_MODE, device_target='GPU', device_id=1)
# 配置全局单位
set_global_units('A', 'kcal/mol')
# 定义一个基于case1.pdb的分子系统
system = Protein('case1.pdb', template=['protein0.yaml'], rebuild_hydrogen=True)
# 定义一个amber.ff99sb的力场
energy = ForceField(system, parameters=['AMBER.FF99SB'])
# 定义一个学习率为1e-03的Adam优化器
min_opt = nn.Adam(system.trainable_params(), 1e-03)
# 定义一个用于执行分子模拟的Sponge实例
md = Sponge(system, potential=energy, optimizer=min_opt)
# RunInfo这个回调函数可以在屏幕上根据指定频次输出能量参数
run_info = RunInfo(200)
# WriteH5MD回调函数,可以将轨迹、能量、力和速度等参数保留到一个hdf5文件中,文件后缀为h5md
cb_h5md = WriteH5MD(system, 'test.h5md', save_freq=10, write_image=False)
# 开始执行分子动力学模拟,运行2000次迭代
md.run(2000, callbacks=[run_info, cb_h5md])
运行结果是这样的:
[MindSPONGE] Adding 45 hydrogen atoms for the protein molecule in 0.003 seconds.
[MindSPONGE] Started simulation at 2023-09-04 15:14:29
[MindSPONGE] Step: 0, E_pot: 1200.4639
[MindSPONGE] Step: 200, E_pot: 7.763489
[MindSPONGE] Step: 400, E_pot: -70.34643
[MindSPONGE] Step: 600, E_pot: -96.88522
[MindSPONGE] Step: 800, E_pot: -109.98717
[MindSPONGE] Step: 1000, E_pot: -117.33747
[MindSPONGE] Step: 1200, E_pot: -121.95378
[MindSPONGE] Step: 1400, E_pot: -125.20764
[MindSPONGE] Step: 1600, E_pot: -127.72044
[MindSPONGE] Step: 1800, E_pot: -129.79828
[MindSPONGE] Finished simulation at 2023-09-04 15:15:16
[MindSPONGE] Simulation time: 46.79 seconds.
--------------------------------------------------------------------------------
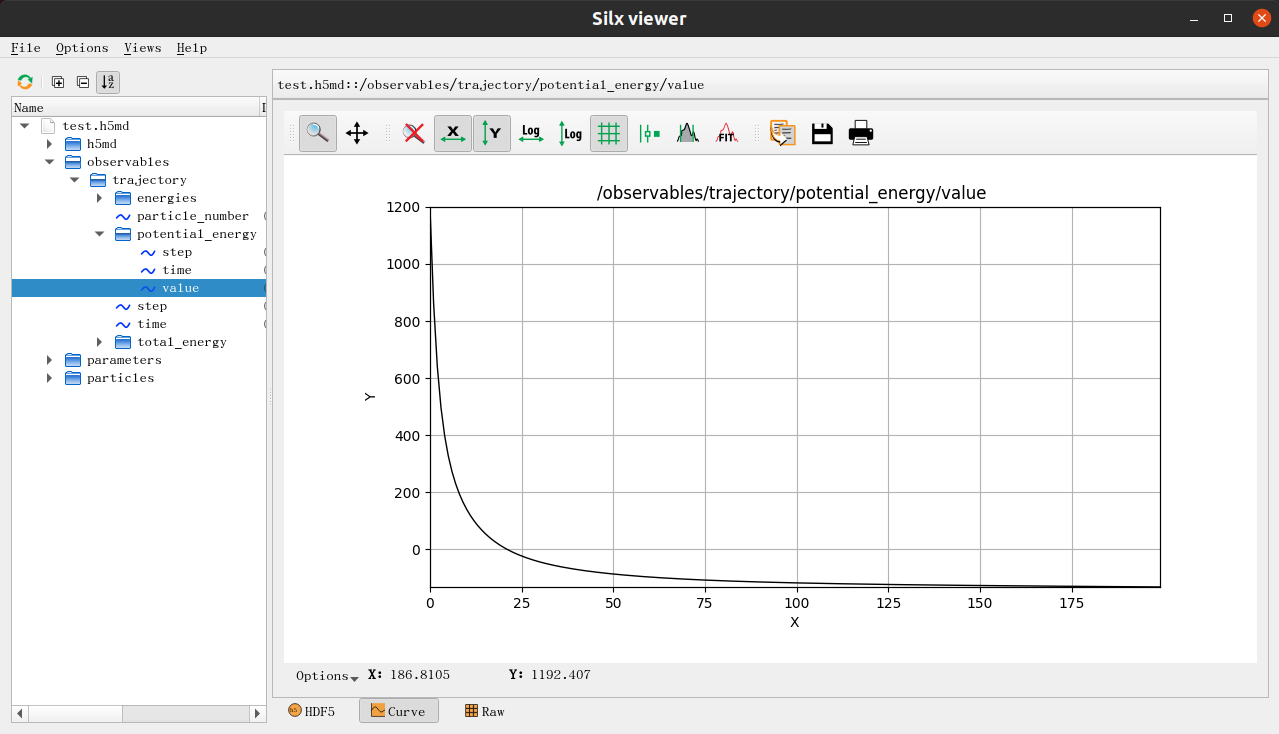
除了在屏幕上输出每一步的能量,我们还通过
WriteH5MD
将整个轨迹写到了一个h5md的文件中,然后可以用silx这样的工具来简单的对h5md文件进行分析和可视化:
定义Collective Variable
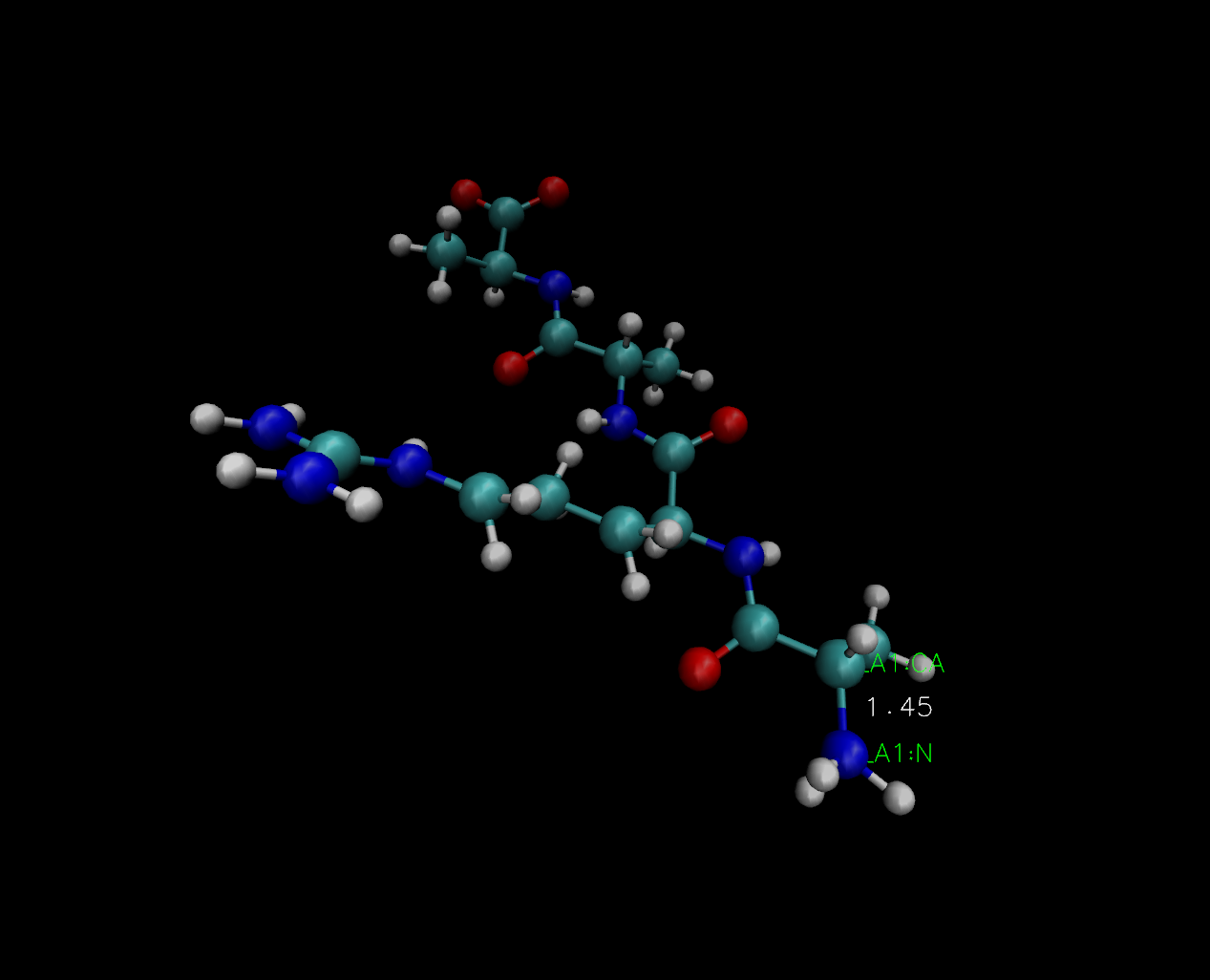
我们在这个体系分子中随便摘取两个原子
\(i\)
和
\(j\)
作为计算CV的参量,首先我们使用一个
距离
作为CV:
\]
如果用VMD来查看这个pdb文件,那么我们定义的CV就是如下图所示的两个原子之间的距离:
在代码侧,我们可以直接从
sponge.colvar
中调用包装好的
Distance
类来定义这个距离的CV。然后作为一个
metrics
传入到
Sponge
对象中,就可以对这个CV的值进行跟踪,作为轨迹的一部分。当然,其实MindSponge也是可以支持使用这个CV去计算一个Bias Energy偏置势的,这就涉及到不同的增强采样算法的不同定义,如Meta Dynamics等,这里暂不做介绍。另外我们还可以基于
sponge.colvar
中的
Colvar
基础类去定义一个我们自己所需要的CV,并传入到metrics中,具体定义形式可以参考
Distance
的写法。
from mindspore import nn, context
import sys
sys.path.insert(0, '../..')
from sponge import ForceField, Sponge, set_global_units, Protein
from sponge.callback import RunInfo, WriteH5MD
from sponge.colvar import Distance
# 配置MindSpore的执行环境
context.set_context(mode=context.GRAPH_MODE, device_target='GPU', device_id=1)
# 配置全局单位
set_global_units('A', 'kcal/mol')
# 定义一个基于case1.pdb的分子系统
system = Protein('../pdb/case1.pdb', template=['protein0.yaml'], rebuild_hydrogen=True)
# 定义一个amber.ff99sb的力场
energy = ForceField(system, parameters=['AMBER.FF99SB'])
# 定义一个学习率为1e-03的Adam优化器
min_opt = nn.Adam(system.trainable_params(), 1e-03)
# 定义CV
bond0 = Distance([0, 1])
print(system.atom_name)
# 定义一个用于执行分子模拟的Sponge实例
md = Sponge(system, potential=energy, optimizer=min_opt, metrics={'bond0': bond0})
# RunInfo这个回调函数可以在屏幕上根据指定频次输出能量参数
run_info = RunInfo(200)
# WriteH5MD回调函数,可以将轨迹、能量、力和速度等参数保留到一个hdf5文件中,文件后缀为h5md
cb_h5md = WriteH5MD(system, 'test.h5md', save_freq=10, write_image=False)
# 开始执行分子动力学模拟,运行2000次迭代
md.run(2000, callbacks=[run_info, cb_h5md])
运行输出如下所示:
[MindSPONGE] Adding 45 hydrogen atoms for the protein molecule in 0.003 seconds.
[['N' 'CA' 'CB' 'C' 'O' 'H1' 'H2' 'H3' 'HA' 'HB1' 'HB2' 'HB3' 'N' 'CA'
'CB' 'CG' 'CD' 'NE' 'CZ' 'NH1' 'NH2' 'C' 'O' 'H' 'HA' 'HB2' 'HB3' 'HG2'
'HG3' 'HD2' 'HD3' 'HE' 'HH11' 'HH12' 'HH21' 'HH22' 'N' 'CA' 'CB' 'C'
'O' 'H' 'HA' 'HB1' 'HB2' 'HB3' 'N' 'CA' 'CB' 'C' 'O' 'OXT' 'H' 'HA'
'HB1' 'HB2' 'HB3']]
[MindSPONGE] Started simulation at 2024-02-19 15:33:57
[MindSPONGE] Step: 200, E_pot: 8.525536, bond0: 1.4885269
[MindSPONGE] Step: 400, E_pot: -70.148384, bond0: 1.4816087
[MindSPONGE] Step: 600, E_pot: -96.796265, bond0: 1.4798437
[MindSPONGE] Step: 800, E_pot: -109.93948, bond0: 1.4798721
[MindSPONGE] Step: 1000, E_pot: -117.30916, bond0: 1.4806045
[MindSPONGE] Step: 1200, E_pot: -121.934845, bond0: 1.4812899
[MindSPONGE] Step: 1400, E_pot: -125.193565, bond0: 1.4817244
[MindSPONGE] Step: 1600, E_pot: -127.70915, bond0: 1.4819595
[MindSPONGE] Step: 1800, E_pot: -129.78864, bond0: 1.482085
[MindSPONGE] Step: 2000, E_pot: -131.60873, bond0: 1.4821533
[MindSPONGE] Finished simulation at 2024-02-19 15:34:44
[MindSPONGE] Simulation time: 47.89 seconds.
--------------------------------------------------------------------------------
可以看到,相比于之前纯粹的能量极小化版本代码,这里我们多打印了一个
bond0
的参数,这里就是每一步优化能量之后对应的CV的值。类似的,我们还可以用
Angle
,
Torsion
等MindSponge内置的CV类型去计算:
from mindspore import nn, context
import sys
sys.path.insert(0, '../..')
from sponge import ForceField, Sponge, set_global_units, Protein
from sponge.callback import RunInfo, WriteH5MD
from sponge.colvar import Distance, Angle, Torsion
# 配置MindSpore的执行环境
context.set_context(mode=context.GRAPH_MODE, device_target='GPU', device_id=1)
# 配置全局单位
set_global_units('A', 'kcal/mol')
# 定义一个基于case1.pdb的分子系统
system = Protein('../pdb/case1.pdb', template=['protein0.yaml'], rebuild_hydrogen=True)
# 定义一个amber.ff99sb的力场
energy = ForceField(system, parameters=['AMBER.FF99SB'])
# 定义一个学习率为1e-03的Adam优化器
min_opt = nn.Adam(system.trainable_params(), 1e-03)
# 定义CV
cv_bond = Distance([0, 1])
cv_angle = Angle([0, 1, 2])
cv_dihedral = Torsion([0, 1, 2, 3])
# 定义一个用于执行分子模拟的Sponge实例
md = Sponge(system, potential=energy, optimizer=min_opt, metrics={'bond': cv_bond, 'angle': cv_angle,
'dihedral': cv_dihedral})
# RunInfo这个回调函数可以在屏幕上根据指定频次输出能量参数
run_info = RunInfo(200)
# WriteH5MD回调函数,可以将轨迹、能量、力和速度等参数保留到一个hdf5文件中,文件后缀为h5md
cb_h5md = WriteH5MD(system, 'test.h5md', save_freq=10, write_image=False)
# 开始执行分子动力学模拟,运行2000次迭代
md.run(2000, callbacks=[run_info, cb_h5md])
运行输出如下所示:
[MindSPONGE] Adding 45 hydrogen atoms for the protein molecule in 0.004 seconds.
[MindSPONGE] Started simulation at 2024-02-19 15:43:11
[MindSPONGE] Step: 200, E_pot: 8.52552, bond: 1.4885279, angle: 1.9996612, dihedral: 2.14814
[MindSPONGE] Step: 400, E_pot: -70.14837, bond: 1.4816078, angle: 1.9796473, dihedral: 2.1302822
[MindSPONGE] Step: 600, E_pot: -96.796265, bond: 1.4798437, angle: 1.9628391, dihedral: 2.1359558
[MindSPONGE] Step: 800, E_pot: -109.939514, bond: 1.4798703, angle: 1.9526237, dihedral: 2.1443036
[MindSPONGE] Step: 1000, E_pot: -117.30916, bond: 1.4806036, angle: 1.9461793, dihedral: 2.1488183
[MindSPONGE] Step: 1200, E_pot: -121.93486, bond: 1.4812908, angle: 1.9418821, dihedral: 2.1499696
[MindSPONGE] Step: 1400, E_pot: -125.19357, bond: 1.4817244, angle: 1.9388888, dihedral: 2.1492646
[MindSPONGE] Step: 1600, E_pot: -127.70906, bond: 1.4819595, angle: 1.9367135, dihedral: 2.1476927
[MindSPONGE] Step: 1800, E_pot: -129.78867, bond: 1.4820842, angle: 1.9350405, dihedral: 2.1457095
[MindSPONGE] Step: 2000, E_pot: -131.60872, bond: 1.4821533, angle: 1.9336755, dihedral: 2.143513
[MindSPONGE] Finished simulation at 2024-02-19 15:44:03
[MindSPONGE] Simulation time: 51.27 seconds.
--------------------------------------------------------------------------------
保存轨迹
在上一步运行之后,会在本地生成一个h5md文件,我们可以用silx view查看这个文件。此时我们会发现,在代码中定义的metrics中几个CV物理量,也会被同步保存到h5md轨迹文件中:

总结概要
随着分子动力学模拟技术的应用推广、AI软件的发展和硬件算力水平的提升,我们可以更快的在分子层面去观察和研究分子体系内的相互作用。但是分子模拟的性能再好,也不一定可以复现一些在自然宏观状态下有可能发生的化学反应或者是物质相变。因此我们需要通过定义一些对反应路径有决定性影响的物理量,然后结合增强采样技术,去更快的复现和推导我们所需要的反应机理。本文主要介绍分子动力学模拟软件MindSponge在这一领域的应用和代码实现。
版权声明
本文首发链接为:
https://www.cnblogs.com/dechinphy/p/cv.html
作者ID:DechinPhy
更多原著文章:
https://www.cnblogs.com/dechinphy/
请博主喝咖啡:
https://www.cnblogs.com/dechinphy/gallery/image/379634.html